Minibinder design is just not that hard [[1]]
Minibinders to PDL1
One computational project we've been working on at Escalante is designing minibinders: small (typically <100 amino acids) protein binders. Minibinders have a huge range of uses from therapeutics to in vitro assay development. There's been a lot of well-justified excitement in the area of computational de novo design of minibinders: it's (almost) fair to say that in the last 24 months de novo minibinder design went from exceedingly difficult and expensive to almost routine (see boolean biotech for a good overview).
While many of these advances have taken the form of closed models trained by large computational teams at private companies, we think open-source models will ultimately win out: there is no real data moat for binder design today [[2]], and, unlike the early days of LLMs, there is little computational moat [[3]]. In fact, the partially open-source package, BindCraft, is one of the top binder design methods. Incidentally, this is pretty clearly a good thing: where would biotech be today if designing a PCR primer required signing a contract with a for-profit company?
In this post we give a short introduction to our open-source protein design package, mosaic, and some preliminary experimental results.
In short, to design a minibinder right now we think you should:
- Absolutely use
BindCraftif you want an experimentally-verified, push-button method for straightforward binder design (and don't mind buying a Rosettta license --FreeBindCraftis an alternative if not). It works. - Try
mosaicif you have a more complicated design objective (dual-target binders, specific folds, lab-in-the-loop, etc), want access to multiple models for design or filtering (Boltz2, AF2, Protenix, etc), or would like to quickly prototype your own models or loss functions for design.
What is mosaic?
At Escalante we often want to optimize additional properties in addition to binding to a single specified target. These properties can range from somewhat standard (e.g. highly expressible and soluble designs following a fixed scaffold fold) to more exotic (e.g. a single minibinder that binds to two very distinct targets). To describe how we generate designs that satisfy these constraints we need to take a quick step back and review how mosaic and BindCraft work.
mosaic and BindCraft formulate the design problem as an optimization problem:
$$\underset{s \in A^n}{\textrm{minimize}}~\ell(s).$$
Here \(A^n\) is the set of all protein sequences of length \(n\) and \( \ell: A^n \mapsto \mathbf{R} \) is a loss functional that scores potential designs. While there are many optimization algorithms one can use to try to solve this problem (generate sequences with small \(\ell(s)\)), BindCraft and mosaic use a clever continuous relaxation for efficiency [[4]].
In the standard BindCraft setup, \( \ell \) is composed of a few terms that are all functions of the output of AlphaFold2, typically something like
$$ \ell(s) = -\textrm{PLDDT}(s) + \textrm{PAE}(s, t) -\textrm{LogContactProb}(s, t) \ldots$$
For those who've used BindCraft this should look familiar, colloquially we're looking for a sequence that AF2 confidently cofolds in contact with the target. But what if we wanted to use models that aren't AF2, or if we wanted to use AF2 more than once?
mosaic simply allows \(\ell\) to be composed of many different properies that can be predicted by entirely different models (e.g. Protenix, ESMC, ProteinMPNN...). Combining these properties into a single composite objective ensures they're taken into account during design. We think this approach is crucial to take advantage of the increasingly-vibrant space of open models for protein property prediction and solve more complicated design problems.
By default we solve these problems using direct optimization, but the same codebase can be used to guide, finetune, or apply reinforcement learning to generative models. We think it's pretty cool!
Preliminary experimental results
As a first test of mosaic in the lab, we designed binders with three different loss functions against two benchmarking targets, PDL1 and IL7RA, using the excellent Adaptyv Bio.
The first objective function we tested is a fairly straightforward reproduction of BindCraft, in mosaic this is something like:
import mosaic.losses.structure_prediction as sp
mpnn = ProteinMPNN.from_pretrained(sol_path)
structure_loss = (sp.BinderTargetContact()
+ sp.WithinBinderContact()
+ 0.05 * sp.TargetBinderPAE()
+ 0.05 * sp.BinderTargetPAE()
+ 0.025 * sp.IPTMLoss()
+ 0.4 * sp.WithinBinderPAE()
+ 0.025 * sp.pTMEnergy()
+ 0.1 * sp.PLDDTLoss())
af2_loss = AlphaFoldLoss(structure_loss)
This is essentially a regression test; while mosaic is inspired by BindCraft, it's a completely new codebase and optimization algorithm.
Next, we wanted to test if adding another model to the loss function could improve design. To that end we added a sequence recovery loss using ProteinMPNN: structures that are easy to inverse fold are often more soluble and stable, and less likely to be "adversarial" unnatural sequences. In mosaic this is only a few lines of code!
mpnn = ProteinMPNN.from_pretrained(soluble_path)
sequence_recovery_loss = 5.0 * SequenceRecovery(mpnn, temp=0.01)
af2_plus_mpnn_loss = AlphaFoldLoss(structure_loss + sequence_recovery_loss)
(okay, there's a bit more boilerplate, check out the repo).
Finally, we wanted to see if swapping AF2 for a more modern open structure prediction model, Boltz-2, would improve our designs. Again, this is very easy to do in mosaic:
boltz2_plus_mpnn_loss = Boltz2Loss(structure_loss + sequence_recovery_loss)
For each of these objectives and each of the two targets, we use the exact same optimization machinery to generate candidate designs, which we then inverse fold, filter, and rank using the standard BindCraft protocol. For each design method and target we sent 10 designs for wet lab testing.
Does this work?
Yes! The top-line numbers are quite good: all three methods produced binders [[5]] for both targets. Our best design method had a success rate of 8/10 for PDL1 and 7/10 for IL7Ra; which is on par with some recently published results from closed models and roughly matches open source work. The strongest binders for each target have affinities in the high nano- to micromolar range; not quite state of the art, but more than good enough for many applications [[6]] – most importantly, we did this with open source tools, without training a single model, and for a total cost of a few thousand dollars (almost all from wet-lab testing).
In terms of our hypotheses above, we can safely conclude mosaic isn't noticeably worse than BindCraft when we use a similar loss function. Similarly, it seems fair to conclude that adding the sequence recovery loss improved designs for these targets. Interestingly Boltz-2 did not perform noticably better than AF2 in these experiments.
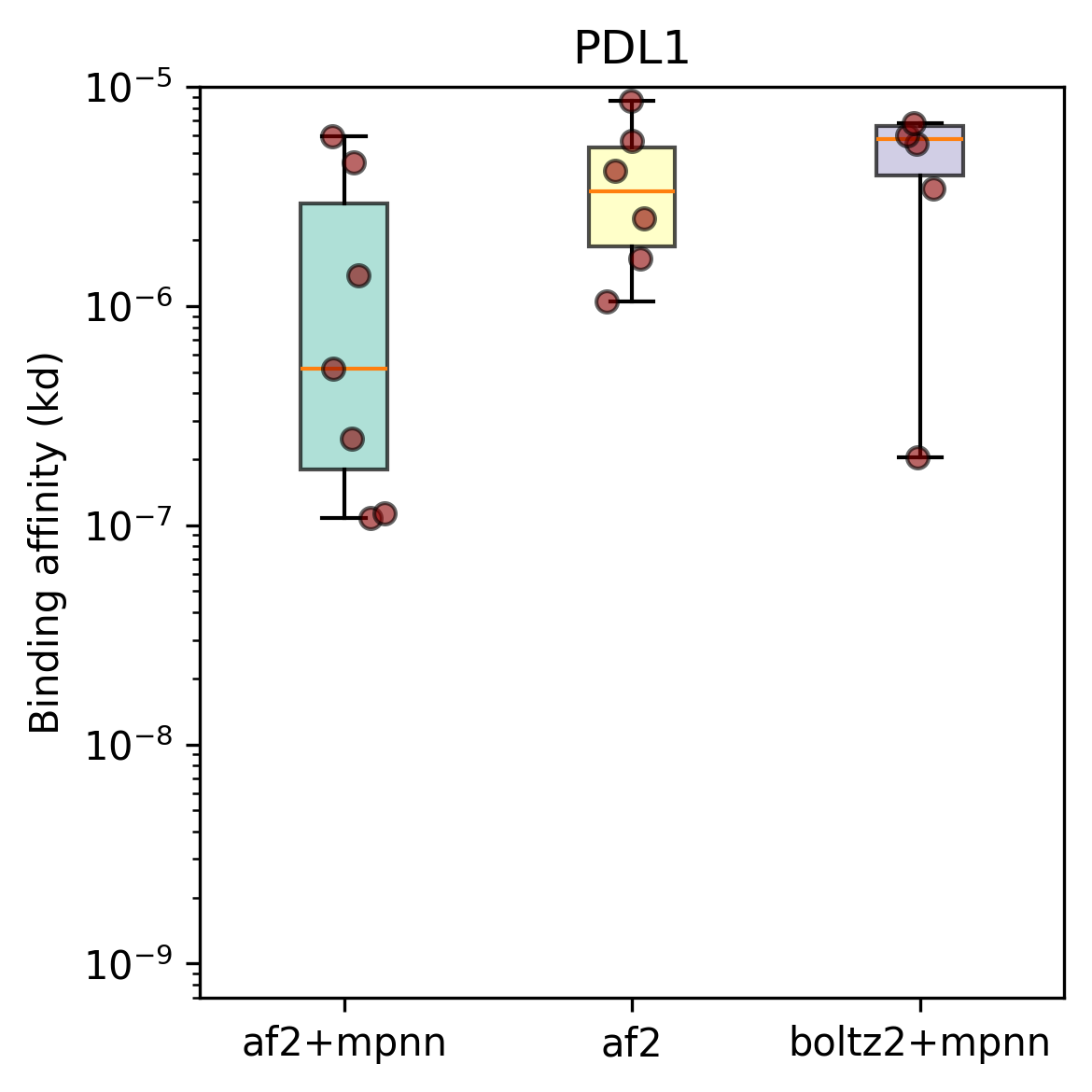
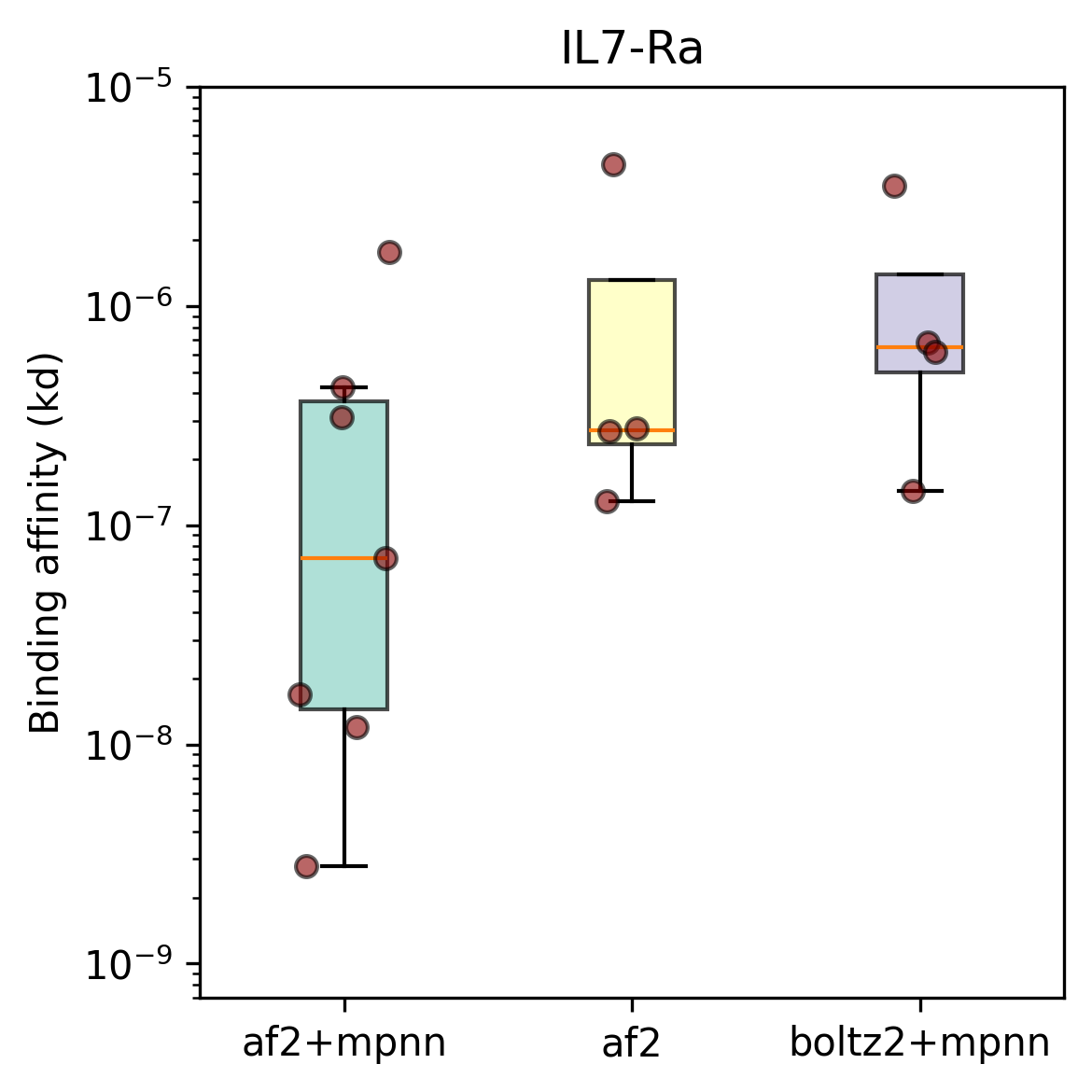
Check out the full results here.
What's next?
There's a lot to do [[7]]!
For this first test we were conservative in our choice of design objectives, we'd next like to try some of the more exotic possibilities that mosaic enables that would be difficult in vanilla BindCraft -- we'll publish more on this soon.
Similarly, the BindCraft filters are clearly excellent, but they're computationally very expensive and rely on Rosetta, which has a non-permissive license; replacing them could speed up design and make this more accessible.
While Boltz-2 underperformed relative to the other methods here, it's not clear if that's due to technical details of our design process (for instance, we first refold with AF2-multimer before running the BindCraft filters), an issue with the weights in the objective function, or the model itself. Further experiments are needed to clarify.
Some very exciting recent work has explored smaller and cheaper structure prediction models, which could substantially speed up design (more on this very soon).
As we hinted at above, replacing direct optimization with generative models (or mixing the two, e.g. via guidance) might be a valuable direction, though it would lose a lot of the flexibility of the pure-optimization approach (more in a future post).
[[1]]: We're far from the first to make this observation (and really, BindCraft proved it), and we're overstating the case: minibinder design for targets like these (well-structured monomers that appear frequently in the PDB) appears to be pretty easy. There are a bunch of interesting extensions of binder design that we think are likely to remain open problems in the near future: designing against more difficult targets and epitopes (including PTMs), with fewer off-target interactions, etc. Also, turning these things into therapeutics is a much, much more difficult problem.
[[2]]: Currently almost everybody trains on the PDB. There are a few companies collecting data for this problem; they'll likely have a short-term advantage in designing better in-silico filters and generative models. To make real progress on binding we think you'd need a huge amount of uniform, high-quality data, at least on the scale of the PDB. Stay tuned. That said there are many non-binding properties where private data absolutely exists and is valuable; for instance anything under the category of developability.
[[3]]: Again, everyone trains on the PDB, which isn't that large. Two trends might add a small barrier here: MD-generated data and perhaps throwing more compute at design and search.
[[4]]: For details, see the readme in our repository.
[[5]]: The minimum detection threshold is probably on the order of 10µM -- not a particularly strong binder, but more than good enough for our use case. As an aside, one reason it's hard to compare different design results is inconsistent detection thresholds between different experimental setups.
[[6]]: Another annoying trend is not reporting how much compute was applied during a design campaign: throwing more compute at the problem before ranking is likely to yield stronger binders. For each of our campaigns here we sampled exactly 2500 designs before ranking. Cost varied based on target and design method, but that works out to at most $200 per target using spot TPU v6e's. Interestingly, more time was spent filtering than designing! Another way to get better affinity results is to make very, very large minibinders. Here, instead, we use relatively short minibinders of 80 AA to fit comfortably within the limits of a 300 NT Twist oligo pool.
[[7]]: mosaic is fully open source, feel free to submit a PR or reach out to collaborate.